Guide
In recent years, research on RNA modification has become one of the most popular research directions in the field of life sciences. With the publication of CNS articles, the molecular mechanism of m5C RNA modification has become more and more clear, and scholars have been searching for more comprehensive. Get the original look of the real m5C modification. Recently, Frank Lyko and Mark Helm from Germany published an article entitled "Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs" at GENOME RESEARCH (IF=11.922). In this paper, they used a comprehensive methylation assay to detect m5C modifications of tRNAs, rRNAs, and mRNAs in mouse embryonic stem cells.
It is pointed out that the technique based on bisulfite conversion is a classical method for detecting m5C modification, and the second generation sequencing provides an opportunity to detect the overall modification map of the transcriptome level. Methods for directly matching m5C RNA modification sites to their native sequences are currently only provided by sulfite-based transcriptome sequencing. (Schaefer et al. 2009). This sequencing principle is selective deamination: converting those unmethylated cytosines (C) to uracil (U) and then revealing methylation-related sequence polymorphisms by second generation.
In this study, the researchers discovered all known tRNA methylation sites and two previously unknown sites that were evolutionarily conserved in 28S rRNA by whole transcriptome sequencing based on bisulfite conversion. In addition, they found that the methylation sites of mRNAs were sparse or completely unmethylated. Researchers say their approach can be used in many experiments to describe the m5C RNA methylation pattern, helping us to better understand the function of m5C RNA methylation in RNA biology and human disease.
1. M5C RNA methylation full transcriptome sequencing based on bisulfite conversion
The researchers established a complete analysis of their own, and subjected to the bisulfite-based m5C RNA methylation transcriptome sequencing of mouse embryonic stem cells. They divided the total RNA sample into two parts, small RNA (<200 nt) and long RNA (>200 nt). A step of removing the rRNA is selectively added based on whether or not the rRNA in the sample is to be detected. The long RNA portion was further fragmented to meet the Illumina sequencing process. Prior to deep sequencing, both fractions of the RNA were treated with DNase, followed by heavy sulfite conversion, and the ends were ligated with the linker for cDNA library construction. After detailed quality control and sequencing comparison database, a series of raw data were obtained. Considering the incomplete conversion of candidate RNA methylation sites, they set up a special set of data filtering conditions. In the methylation peak calling phase, each sample is subjected to a Poisson distribution and the non-conversion rate p value is calculated. Finally, three biological replicates were calculated and a comprehensive non-conversion rate p-value was calculated.
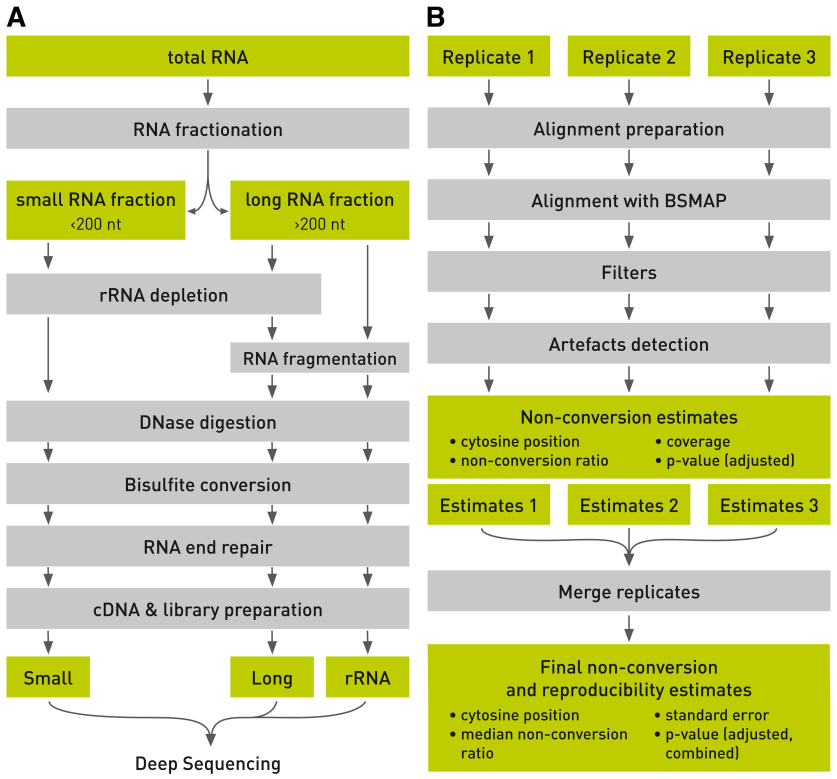
Figure 1: Schematic diagram of m5C RNA methylation full transcriptome sequencing based on bisulfite treatment
2. Perform mRNA-related methylation site analysis on the first library raw data
The researchers found that most of the cytosine conversions were >90%. To further analyze those untransformed cytosine reads, they set a Poisson parameter λp, which is counted in each coverage layer (10X-1200X). The count of untransformed cytosine Nn; and calculated a ratio r = λp / (Nn + Nc), where Nc is the converted cytosine count and N = Nn + Nc is the coverage layer. In the subsequent steps, they compared the basal distribution of each cytosine site with non-conversion and non-conversion rates above λp/coverage. Of the 3,338,384 cytosines, 53,510 unconverted rates were greater than or equal to 0.2, which is consistent with previous findings. However, after Benjamini-Hochberg corrected multiple tests, only 266 of the 53,510 cytosines were p<0.05. A large number of data that do not match the p-value indicate that statistical methods are needed to measure the entire transcript histidine subunit sequencing data set. Their Poisson parameter method was affirmed after the non-conversion rate data was filtered on the three biological replicates.
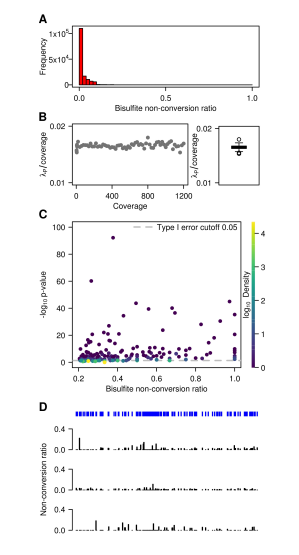
Figure 2: Data analysis of sequencing results of mouse ES cell mRNA
3. Further measurement of Poisson test data analysis
To assess the type I error of this statistical analysis method, they also evaluated the proportion of false positives produced by the Poisson test by simulating hundreds of random untransformed cytosine examples. The result is that this set of analytical methods reliably (statistical power = 99.9 ± 0.04%) detects sites with non-conversion rates below 20X as low as 20%. In their sequencing data set, >50% of the cytosine residue coverage > 20X, thus showing sufficient statistical power.
However, although the Poisson test method was followed, some candidate sites could not be considered as true methylation sites. To further validate the reproducibility of candidate methylation sites, they selected 10 candidate sites with minimal normalized p-values ​​and 4 additional candidate sites, their methyl groups. The level of chemistry is close to 1.0 and the p-value reaches a significant standard. These 14 sites were used for amplicon resequencing. The results showed that 4 of these 14 sites did not undergo methylation modification. This again demonstrates that there is a very low number of candidate methylation sites with some false positives.
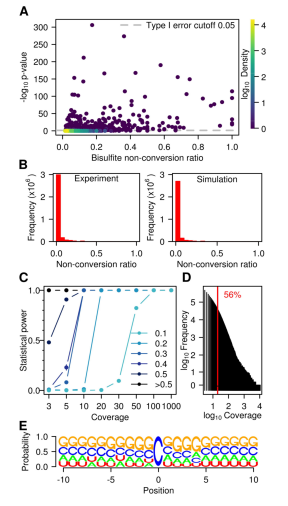
Figure 3: Further data analysis
4. LC-MS/MS detects the overall methylation level of mRNA
Next, the researchers used LC-MS/MS technology to detect the overall methylation level of mRNA. Total RNA was enriched for mRNA in two consecutive rounds of polyA-seleciton, and small RNA was removed, and rRNA was removed in two rounds. Each step was sampled for RNA-seq to see the composition of the three RNAs and the base changes were viewed by LC-MS/MS. The results of RNA-seq show that this shunt process enriches more and more mRNA. However, in the last step of enrichment, 7.1% of rRNA was still detected, probably due to incomplete rRNA fragment removal. The results of LC-MS/MS indicated that the m5C modification was drastically reduced as the enrichment progressed and was closely related to the rRNA removal observed by sequencing. In addition, they did not detect any mRNA 5-hydroxymethylation modifications. The same test was performed using Drosophila S2 cells, and similar results were obtained. This is clearly contrary to previous studies. The results of this experiment also revealed that the m5C methylation modification of mRNA is very sparse or even completely unmethylated.
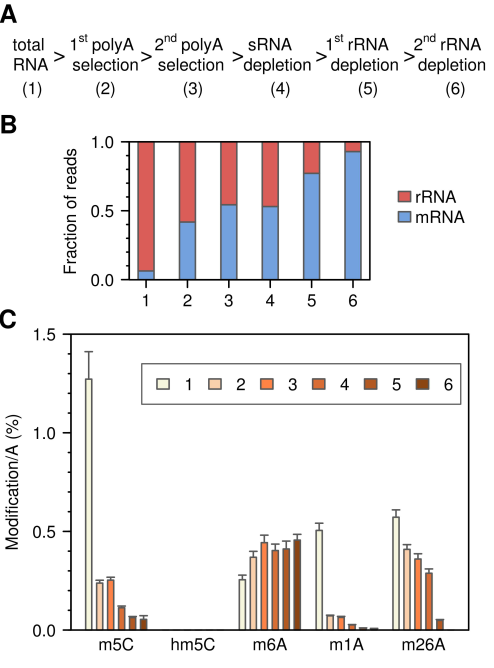
Figure 4: LC-MS/MS detects the overall methylation level of mRNA
5. LC-MS/MS detects the overall methylation level of mRNA
Next, the researchers used LC-MS/MS technology to detect the overall methylation level of mRNA. Total RNA was enriched for mRNA in two consecutive rounds of polyA-seleciton, and small RNA was removed, and rRNA was removed in two rounds. Each step was sampled for RNA-seq to see the composition of the three RNAs and the base changes were viewed by LC-MS/MS. The results of RNA-seq show that this shunt process enriches more and more mRNA. However, in the last step of enrichment, 7.1% of rRNA was still detected, probably due to incomplete rRNA fragment removal. The results of LC-MS/MS indicated that the m5C modification was drastically reduced as the enrichment progressed and was closely related to the rRNA removal observed by sequencing. In addition, they did not detect any mRNA 5-hydroxymethylation modifications. The same test was performed using Drosophila S2 cells, and similar results were obtained. This is clearly contrary to previous studies. The results of this experiment also revealed that the m5C methylation modification of mRNA is very sparse or even completely unmethylated.
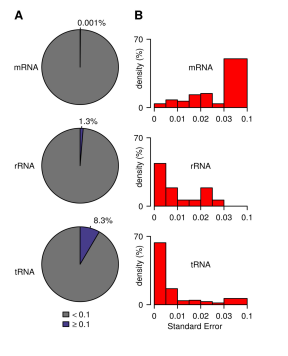
Figure 5: Comparison of methylation levels of three mRNAs: mRNA, rRNA and tRNA
6. Using Poisson test to detect the effects of m5C-related methylase
The researchers finally examined the specificity of RNA m5C enzyme. Based on newly developed computational processes, provide standard analysis of specific types of RNA and compare methylation patterns between different genotypes. They determined the effect on tRNA, rRNA and mRNA methylation levels in the presence or absence of tRNA methyltransferase DNMT2. The results showed that DNMT2 is a highly specific enzyme, and a set of data for DNMT2 mutations showed deletion of methylation at the C38 site in tRNA (Asp), tRNA (Gly) and tRNA (Val). Analysis of rRNA revealed the presence of two new fully methylated cytosines, namely C3438 and C4099, in 28S rRNA.
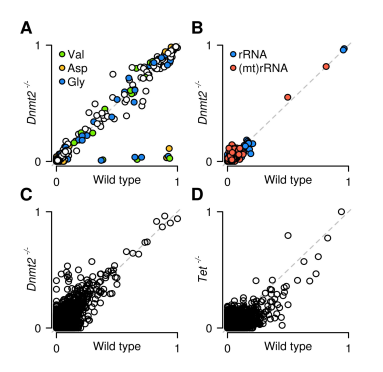
Figure 6: Scatter plot showing untransformed cytosine of tRNA (A), rRNA (B), mRNA (C) in wild-type and DNMT2 knockout mouse ES cells
Summary: This paper identified the most reliable method for whole transcriptome bisulfite sequencing for m5C RNA methylation single base analysis, and showed that there are huge differences in m5C methylation patterns between tRNA, rRNA and mRNA, predicting their exercise with different RNAs. Specific physiological functions are closely related.
About the Author:
Professor Frank Lyko, Ph.D., works at the Epidemiology Group of the German Cancer Research Center (DKFZ) at the University of Heidelberg, Germany, and publishes articles on cell biology and tumor biology. His main research direction is leukemia. Since 1995, a total of 92 sci articles, 19 reviews, 2 short surveys, and 2 letters have been published, totaling 8719 times, mainly related to the apparent modification of DNA and RNA, methylation modification and methylation transfer. Enzymes, etc. He was awarded the Novartis Basic Pharmacology Research Award.
References: Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs
Cloud order related recommendation  Â
m6A RNA methylation sequencing
m5C RNA methylation sequencing
Whole transcriptome sequencing

Shanghai Yunxu Biological Technology Co., Ltd. Â
Shanghai Cloud-seq Biotech Co.,Ltd       Â
Address: 6/F, Building 71, Lane 1066, Qinzhou North Road, Caohejing High-tech Development Zone, Shanghai Â
phone
Website: Â
mailbox:
Application: used in AKD Emulsifier
Product Type: Cassava starch imported from China
Chemical composition: Tapioca starch
Appearance:White powder
Moisture ( % ): ≤ 14.0
Spots ( a / cm2): ≤ 2
Viscosity: 20 ~ 50 s (20%, 40 °C, 4 # Tu's cup)
pH value: 6.0 ~ 7.5
Toxicity: No harm to human health during operation and use
Storage: Store in a cool , dry, ventilated warehouse
Nature: white, gelatinization in low temperature , good fluidity , viscosity stability, aging resistance , the film is good , strong bond , fast drying . Products with low protein content , gelatinization liquid not perishable , no foam , no smell. Good adhesion properties of various fibers , high compatibility with other additives . For AKD emulsifier, the rational use of the product can effectively ensure emulsifying effect and reduce production cost.
Dosage: Production of one ton of 25% emulsifier simply add 192kg of the product
Packing: 25 kg / bag.
|
Product Introduction : |
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||

Akd Special Starch,Oxidized Starch,Starch For Akd Emulsifer,Cassava Starch
Shandong Tiancheng Chemical Co., Ltd. , https://www.tianchengchemical.com